BDB-Lab members involved


AEMB: a computationally efficient abundance estimation method for metagenomic binning
AEMB: a computationally efficient abundance estimation method for metagenomic binning
by Shaojun Pan, Ivan Tolstoganov, Kristoffer Sahlin, Marcel Martin, Xing-Ming Zhao, Luis Pedro Coelho
Abstract
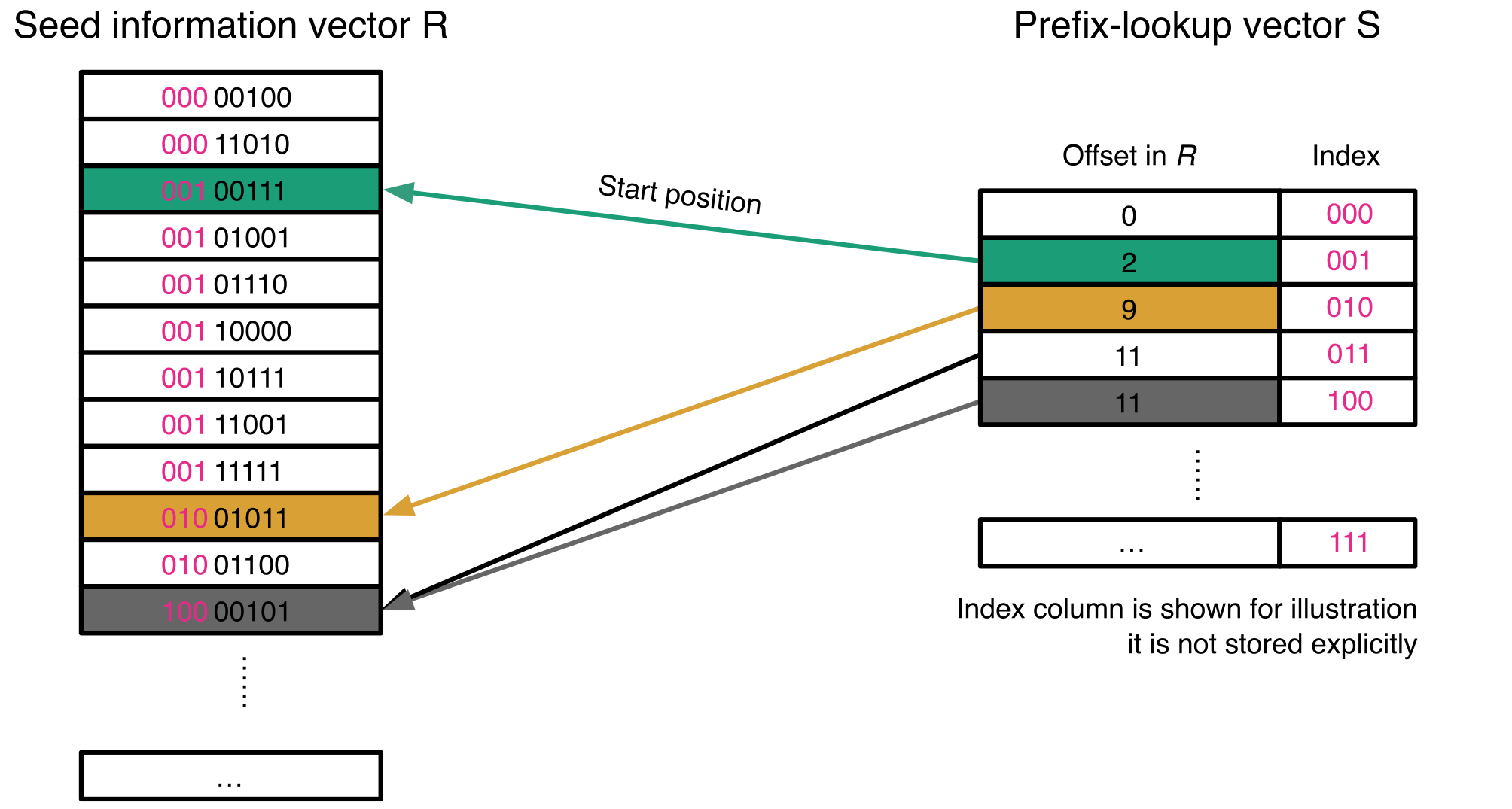
Metagenomic binning is a crucial step in metagenomic analysis, namely grouping together contigs that are predicted to originate from the same genome to enable the recovery of metagenome-assembled genomes (MAGs). It has been shown that using information from multiple samples yields better results than binning each sample independently. However, for N metagenomic samples, using full multi-against-multi binning requires N² alignments, making it computationally challenging to apply in large-scale metagenomic studies. Here, we propose AEMB (Abundance Estimation for Metagenomic Binning), a novel mapping mode implemented in strobealign. AEMB is a computationally efficient abundance estimation method that uses a prefix-lookup vector as an indexing structure to reduce memory usage and randstrobes to estimate the abundance of contigs without performing base-level alignment. Compared to the hash table used in the previous version of strobealign, the indexing structure reduces peak memory usage by 25.2% with almost the same runtime. Furthermore, we implemented a fast abundance estimation method that skips base-level alignment. Altogether, AEMB reduces the runtime for abundance estimation by 88% to 96% compared to commonly used alignment methods such as Bowtie2 and BWA, while achieving similar binning results. AEMB is available as a mapping mode in strobealign https://github.com/ksahlin/strobealign and SemiBin2 (v2.1 and later) accepts its inputs for binning.
Full text: https://doi.org/10.1101/2025.07.30.667338
Copyright (c) 2018–2026. Luis Pedro Coelho and other group members. All rights reserved.